

MedTech Development Regulatory Support
UofG MedTech development landscape includes a wide range of medical products such as surgical instruments, software, hardware, firmware, scientific instruments and in-vitro diagnostics.
Developing new technologies involves navigating various regulatory requirements to ensure they can be safely used in patients and approved for market authorisation in the future.
To help address these challenges, the CETC developed resources to support UofG MedTech innovators in navigating the MD regulatory pathway during the early stages of product development.
These CETC resources should be used alongside informal regulatory guidance and formal scientific advice from relevant regulatory bodies including the UK (MHRA), EU (EMA) and US (FDA) as applicable.
CETC MedTech Regulatory Support
- Medical device regulatory educational platform RegMetrics
- Regulatory Strategy Roadmap
- Guidance and signposting to resources for early phase development of a Quality Management System (QMS) framework and Technical File (TF)
- Help identify and engage with suitable external regulatory consultants
For support and signposting to resources related to MedTech regulatory input, please contact the CETC Innovation Manager directly. Or book drop-in session.

1. Medical device regulatory educational platform - Reg Metrics
RegMetrics is an educational platform for Medical Device Regulation. It is widely used by translational research services at universities and hospitals, as well as by MedTech incubators and accelerators to assist entrepreneurs in navigating their regulatory journey.
The software supports innovators by helping them determine if their device is a medical device, identify its classification, understand which regulatory requirements apply, discover useful standards, and find appropriate test houses for assistance.
CETC secured the RegMetrics licence with the Translational Research Initiatve (TRI) funding and is available to any individual with a UofG email address until April 2026 in the first instance.
Please create your UofG RegMetrics account here: https://www.reg-metrics.com/

2. Regulatory Strategy Roadmap
To establish the regulatory strategy, one first needs to confirm that a product is classified as an MD. Once confirmed, the next step is to determine its risk classification and the appropriate conformity procedure that outlines a streamlined workflow that summarises the key activities and considerations for the development, evaluation and commercialisation of MDs, aiming to facilitate their effective translation into clinical practice.
- Regulatory landscape
- Is my product a medical device?
- What is the classification of the device?
- Conformity procedures
- Essential Requirements (ERs) and General Safety and Performance Requirements (GSPRs)
- Standards
- The Technology Readiness Levels and definitions
Regulatory Strategy Roadmap outlines a streamlined workflow that summarises the key activities and considerations for the development, evaluation and commercialisation of MDs, aiming to facilitate their effective translation into clinical practice. The following sections explain the steps involved in MedTech development and the support available from CETC at each stage of the process.
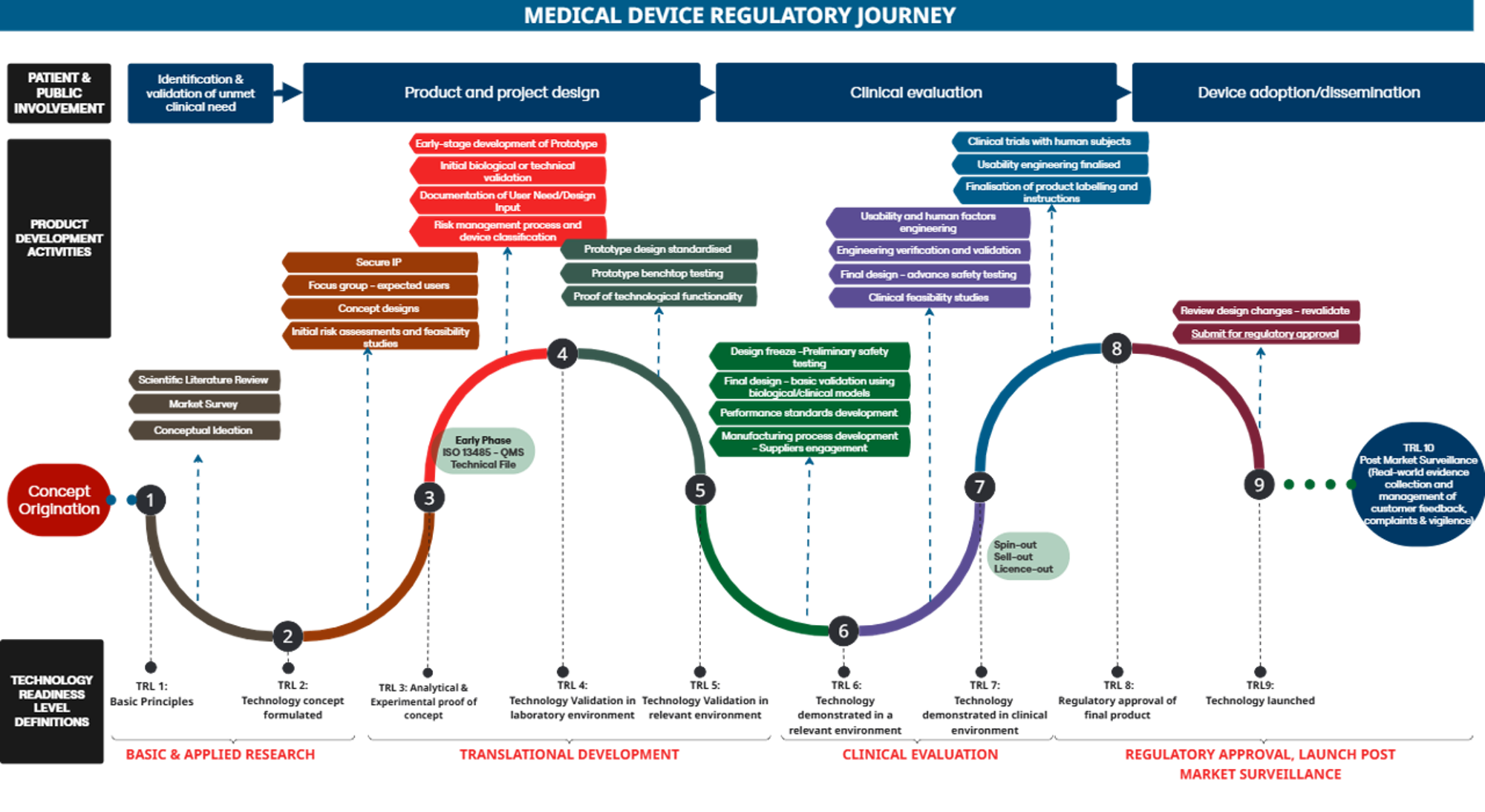

2.1 MedTech Product Design and Development (TRL 1-4)
In the early stages of MedTech development TRL 1-4, the UofG innovator should clearly define the Unmet Clinical Need and Intended Use and initiate Risk Identification & Management Processes
- 2.1.1 Idea and Unmet Clinical Neeed
- 2.1.2 Intellectual Property (IP)
- 2.1.3 Evidence Generation aat the early MedTech development stage (preliminary research, clinical engagement, documentation)

2.2 Laboratory & Pre-clinical Technology Evaluation (TRL 5-6)
In the early research and development phase of a MedTech product TRL 5-6, it is crucial to determine if the research prototype meets all the essential safety and performance requirements. Additionally, it is important to identify the type of research data needed to satisfy the appropriate regulators regarding the appropriate systematic development and testing of the device in accordance with applicable MD legislation.
- 2.2.1 Evidence Generation at the pre-clinical stage of MedTech development (Patient, clinicians, health economics and regulatory engagement, prototype evaluation)

2.3 Clinical Evaluation and Clinical Investigation (TRL 7-8)
- 2.3.1 Clinical Trial Product
- 2.3.2 Clinical evaluation
- 2.3.3 Clinical investigation
- 2.3.4 Important PPI considerations
- 2.3.5 Documentation
- 2.3.6 Financial Implications
- 2.3.7 Regulatory Approval for Clinical Trials

2.4 Commercial Adoption (TRL 9-10)
For the new medical device to progress beyond its primary clinical and performance evaluation (TRL 9-10), it requires commercial adoption outside of the UofG for NHS uptake. Before a product can receive market approval in the UK or Europe, it must be UKCA or CE marked. This certification confirms the product's compliance with health, safety, and environmental protection standards. Holding a patent on a device is effectively meaningless without a UKCA or CE mark.
For more information on the UKCA and CE Mark please refer to the “Regulatory Roadmap for UofG MedTech Developers “ guideline (link to GUI_85.001 here).
The University does not possess CE marks. Therefore, at this stage in the medical device lifecycle, the product transitions from being an 'in-house' development project. In practice, the resources needed for such an undertaking often exceed what UofG can provide, making commercial adoption essential.
Commercial adoption can occur through one of the following methods:
- Sold-out: The device is sold entirely to a company or manufacturer in a one-time deal
- Licensed-out: UofG retains ownership of the device or its components but grants a license to a company for marketing.
- Spin-out: the UofG innovator establishes a separate commercial entity to market the device

3. Early phase development of a Quality Management System (QMS) framework and Technical File (TF)
Innovators at the UofG who are in the early stages of MedTech development often concentrate their limited resources on the technical aspects of product development. Due to constraints in funding and manpower, there is a tendency and necessity to prioritise immediate product functionality over robust quality management practices. This approach can lead to significant challenges later, particularly when it comes to meeting regulatory requirements and ensuring product safety and efficacy
3.1 Quality management system (QMS)

4. Help identify and engage with suitable external regulatory consultants
Regulatory advice is essential for MedTech development and marketing authorisation. Please note that this regulatory input has financial implications and should be planned earlier in research grant applications.
For a list of resources related to available external regulatory consultants and guidance on identifying required estimated funds to help with the development of a Quality Management System (QMS) framework and a Technical File, please contact CETC directly.